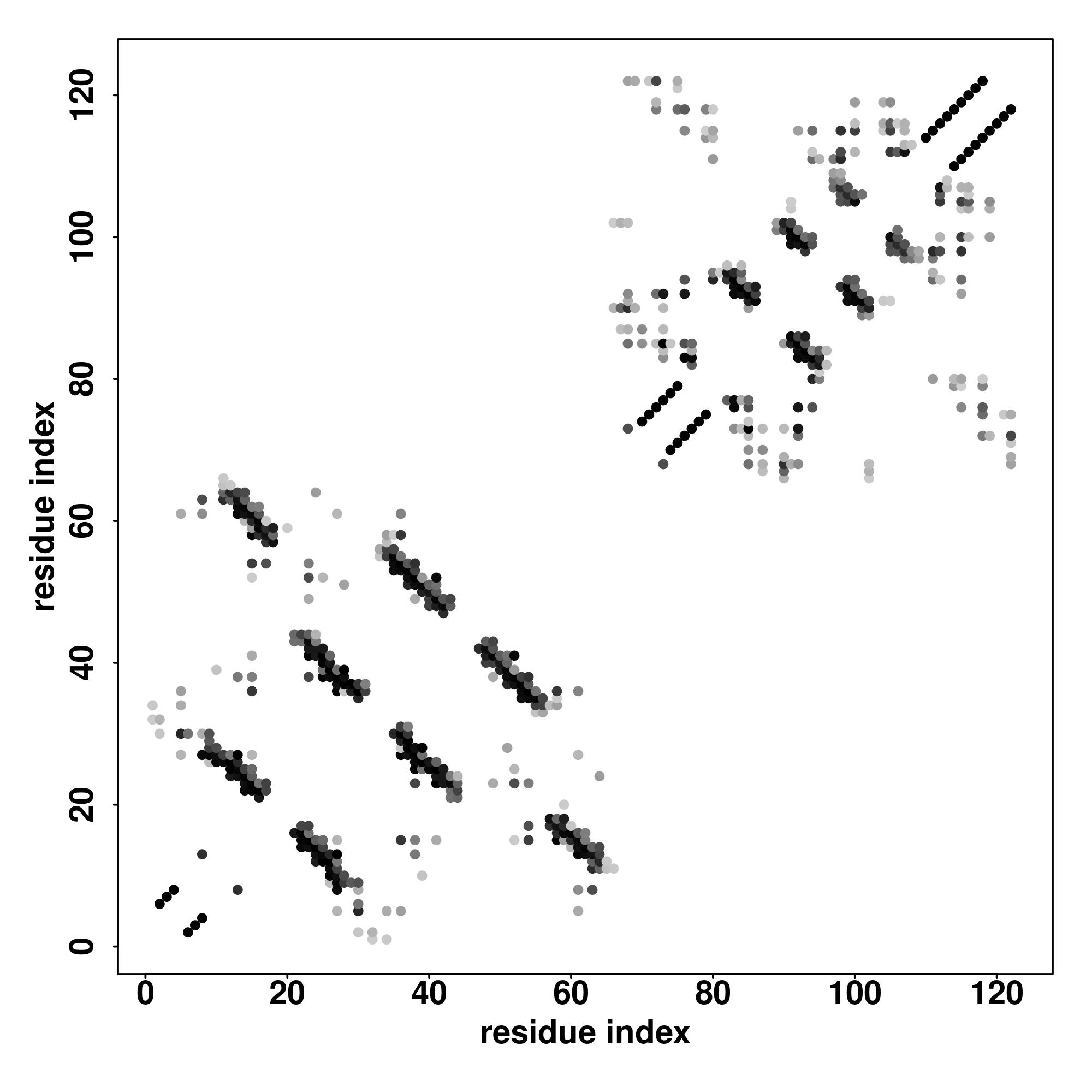
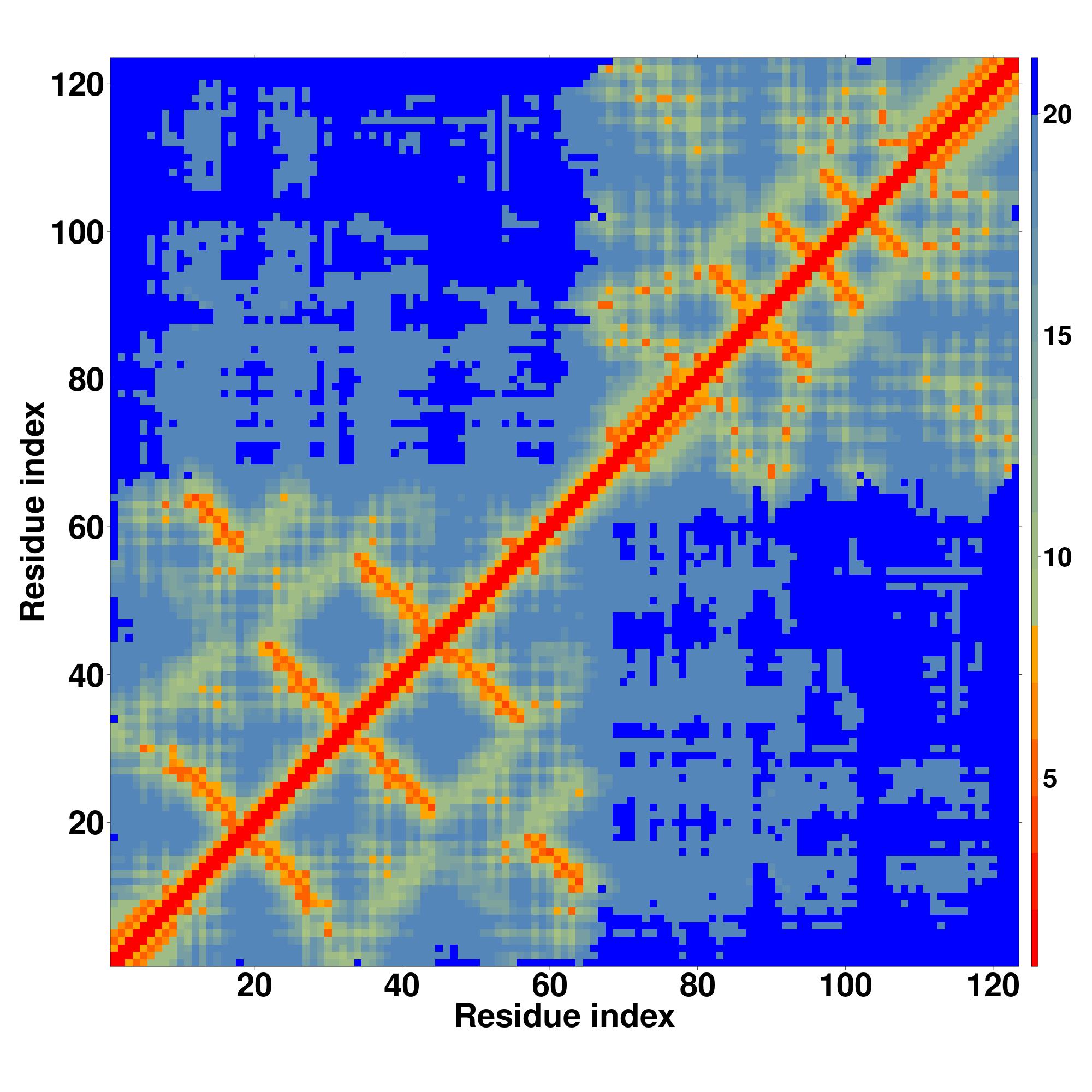
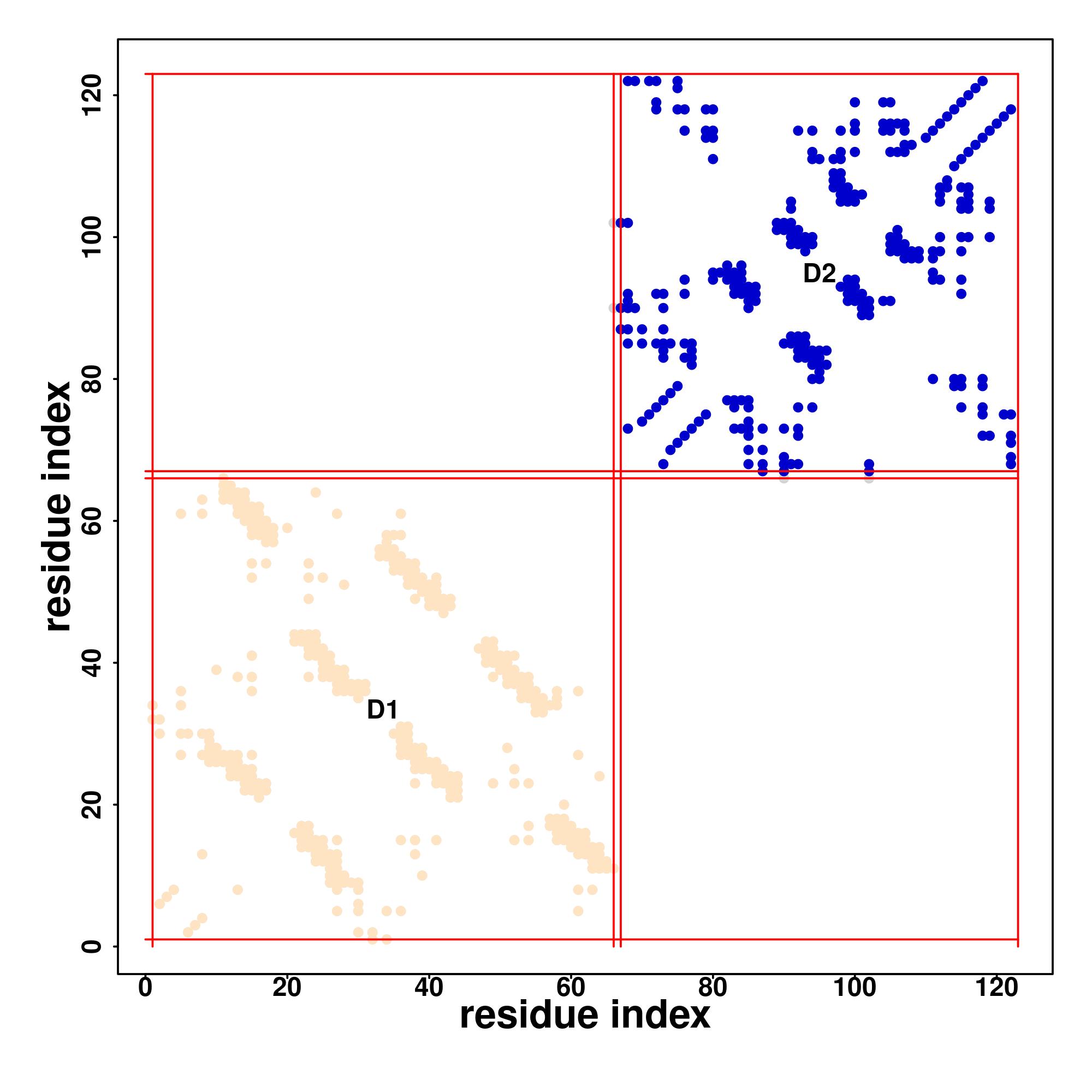
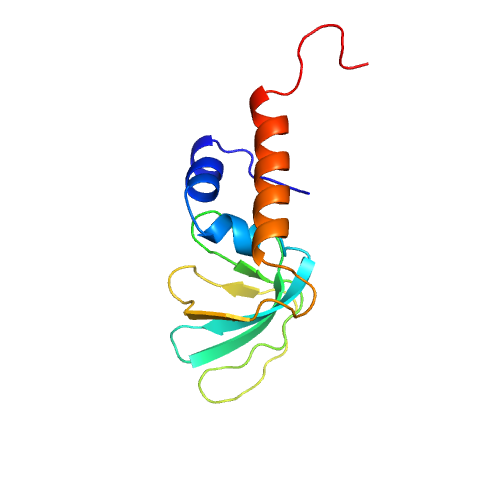
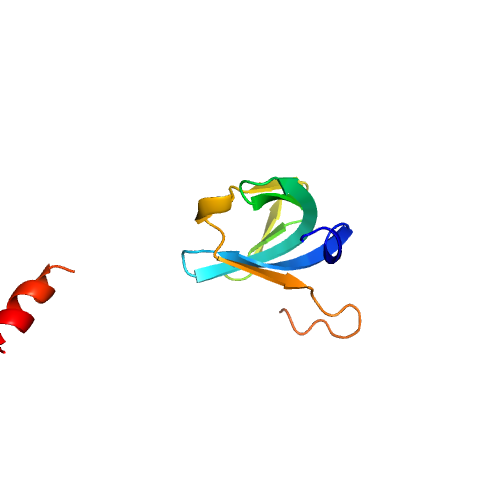
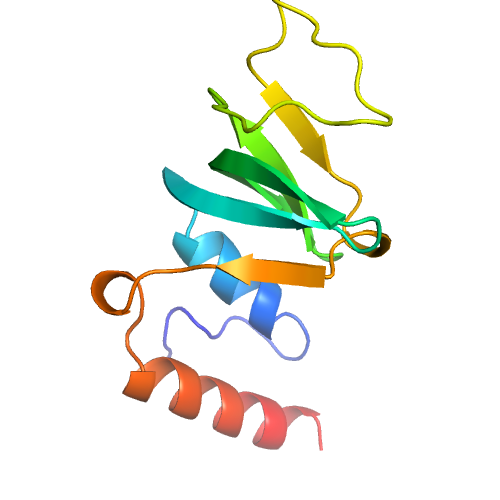
| Rank |
PDB
Hit |
RCSB
Link |
CMO |
MAE |
ID1 |
ID2 |
Cov |
Norm.
Zscore |
Download PDB |
Download FASTA |
Gene_Ontology_(GO)_term
(Molecular_Function) |
Gene_Ontology_(GO)_term
(Biological_Process) |
Gene_Ontology_(GO)_term
(Cellular_Componenet) |
Enzyme_Commission
(EC)_number |
Ligand |
Binding site residues |
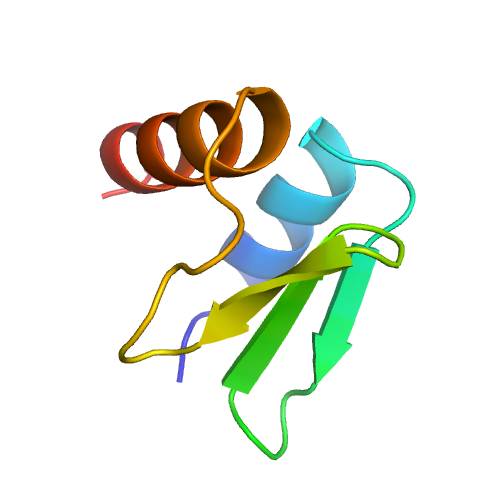
| 1 | 5nrll | 5nrl | 0.454 | 2.064 | 0.18 | 0.14 | 0.80 | 1.31 | template_1 | template_1 | GO:0003723,GO:0003729 | GO:0000387,GO:0006397,GO:0008380,GO:0000398 | GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0005634,GO:0005829,GO:0046540,GO:0071004,GO:0030532 | | | |
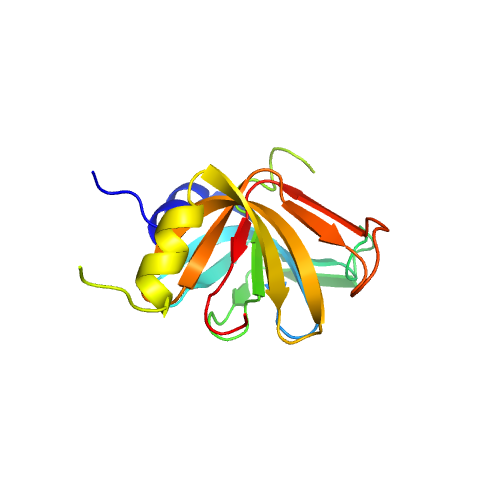
| 2 | 4m77A | 4m77 | 0.398 | 2.325 | 0.20 | 0.13 | 0.67 | 1.18 | template_2 | template_2 | GO:0003723,GO:0005515 | GO:0000398,GO:0016070,GO:0006397,GO:0008380,GO:0006364,GO:0008033 | GO:0005688,GO:0046540,GO:0005634,GO:0005681,GO:0005737,GO:0005730 | | | |
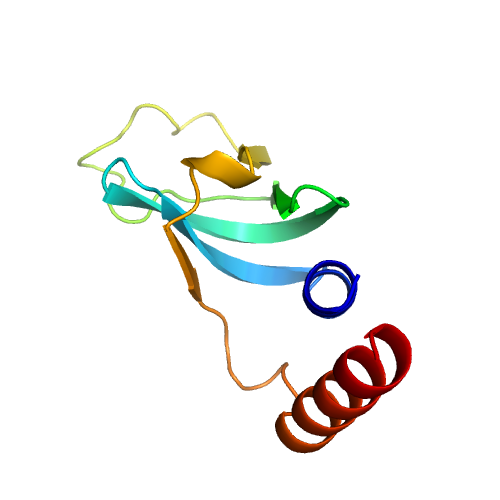
| 3 | 5mqfg | 5mqf | 0.426 | 2.374 | 0.20 | 0.12 | 0.67 | 1.15 | template_3 | template_3 | GO:0003723,GO:1990446,GO:0005515 | GO:0000398,GO:0000387,GO:0006397,GO:0008380,GO:0000245,GO:0051170 | GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0005634,GO:0005681,GO:0005737,GO:0005829,GO:0034709,GO:0046540,GO:0071005,GO:0071007,GO:0005654,GO:0030532 | | | |
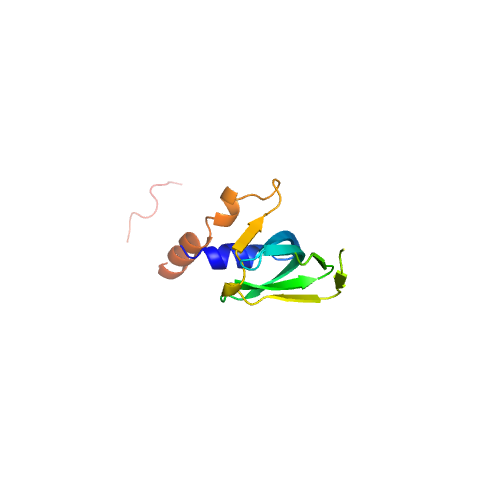
| 4 | 5ganl | 5gan | 0.373 | 2.207 | 0.22 | 0.15 | 0.74 | 1.14 | template_4 | template_4 | GO:0003723,GO:0003729 | GO:0000387,GO:0006397,GO:0008380,GO:0000398 | GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0005634,GO:0005829,GO:0046540,GO:0071004,GO:0030532 | | NUC
| K2 K8 K9 R11 K20 S33 P34 Q35 N37 R61 G62 N63 R66 L79 D82 Q83
|
| 5 | 4c8qB | 4c8q | 0.401 | 2.067 | 0.26 | 0.18 | 0.85 | 1.13 | template_5 | template_5 | GO:0003723,GO:0005515 | GO:0006397,GO:0008380,GO:0006364,GO:0008033,GO:0000398 | GO:0000932,GO:0005688,GO:0046540,GO:1990726,GO:0071013,GO:0005634,GO:0005681,GO:0005737,GO:0005730,GO:0005732 | | CO
| E28 R78 Y79
|
| 6 | 5mqfe | 5mqf | 0.407 | 2.303 | 0.20 | 0.12 | 0.68 | 1.11 | template_6 | template_6 | GO:0003723,GO:0071209,GO:0070034,GO:1990446,GO:0019899,GO:0005515,GO:0071208 | GO:0000398,GO:0000387,GO:0006397,GO:0008380,GO:0006479,GO:0006369,GO:0008334,GO:0051170 | GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0005634,GO:0005681,GO:0005737,GO:0005829,GO:0005654,GO:0005683,GO:0005697,GO:0016604,GO:0034709,GO:0046540,GO:0071005,GO:0071007,GO:0030532 | | | |
| 7 | 3jb9D | 3jb9 | 0.407 | 2.077 | 0.18 | 0.13 | 0.78 | 1.09 | template_7 | template_7 | GO:0003755,GO:0003723,GO:0005515,GO:0003729 | GO:0000413,GO:0006457,GO:0000395,GO:0006396,GO:0000387,GO:0006397,GO:0008380 | GO:0005681,GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0071004,GO:0005634,GO:0046540,GO:0071014 | | NUC
| N35 N37 R61 G62 K81 P84 R96
|
| 8 | 4c8qD | 4c8q | 0.417 | 2.271 | 0.31 | 0.19 | 0.66 | 1.07 | template_8 | template_8 | | GO:0006396,GO:0000398,GO:0000956 | | | | |
| 9 | 5nrld | 5nrl | 0.438 | 2.286 | 0.21 | 0.13 | 0.67 | 1.07 | template_9 | template_9 | GO:0003723,GO:0005515,GO:0003729 | GO:0000387,GO:0006397,GO:0008380,GO:0000398 | GO:0005686,GO:0005685,GO:0000243,GO:0005682,GO:0005687,GO:0005689,GO:0034715,GO:0034719,GO:0071013,GO:0005634,GO:0005737,GO:0046540,GO:0071004 | | | |
| 10 | 5mkl11 | 5mkl | 0.485 | 2.469 | 0.23 | 0.23 | 1.21 | 1.06 | template_10 | template_10 | | | | | | |