
STRUM is a method for predicting stability changes (ΔΔG) of a protein upon single-point mutation.
STRUM adopts a gradient boosting regression approch using variety of features at different levels of evolutionary information and structural resolution (Figure 1).
The unique features of STRUM are the inclusion of some sequence profile scores combining different methods of multiple sequence allignment, some strucutral profile scores reflecting the likelihood of a given amino acid or other properties at mutant position being found in the ensemble of structurally similar protein, and different energy functions based on
I-TASSER model providing accurate environment information.
All the features of STRUM can be generated only from protein sequence, so its ability to deliver good predictions without experimental stucture high-quality extend the application of stability change prediction.
Comparing with several stat-of-the art methods on the 2402 common mutations, the Pearson's correlation coefficient of STRUM between the predicted and measured ΔΔG has been increased to 0.77 from average level 0.66, and RMSE has been reduced to 0.93 from average level 1.10.
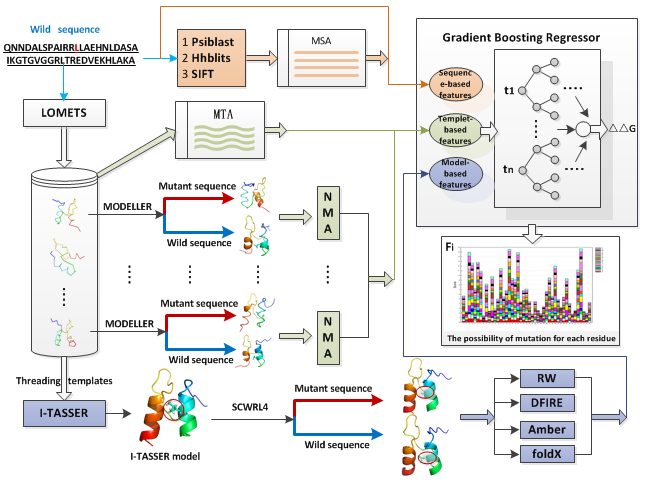 Figure 1
Figure 1: Summarizing the STRUM predictive workflow can be divided into the following steps: First, we select some physicochemical
properties such as volume, molecular weight, hydrophobicity, isoelectric, PSSM and conservation and structural information such as
secondary structure, backbone torsion angles and solvent accessibility for wild-type residue. These information are derived from
sequence as sequence-based features (orange region); Second, query sequence is threaded by
LOMET
though a non-redundant template library to identify homologous and/or analogous structure templates. So a Multiple Template Alignment (MTA) can be
obtained and induce a score related to BLUSUM62 and
TM-score.
Then we use Modeller to rebuild wild-type and mutant structures respectively depended on the wild-type and mutant sequences with each template.
The fluctuations and root mean square innerproduct are calculated based on the wild-type and mutant structure with normal mode analysis (NMA).
These features are classified as template-based features (resedue region). Third, the 3D structure of protein is predicted by
I-TASSER based on iterative Monte Carlo simulation.
The mutant structure was built by SCWRL4. Then two empirical force field potential methods such as Amber and FoldX and two knowledge-based potential methods such as RW and DFIRE were used to generate different energetic features as I-TASSER model-based features (blue-violet region).
Finally, STRUM is trained and tested by using gradient boosting regressor.
To take into account the change in amino acid types due to the mutation, each kind of features is performed for the wild type and mutant residue,
where the blue arrow is related to wild-type information, and the red arrow is related to mutant information.
References:
- Lijun Quan, Qiang Lv, and Yang Zhang.
STRUM: Structure-based stability change prediction upon single-point mutation,
Bioinformatics, 2016: btw361.
(download the PDF file)
