Back to CEthreader homepage
About CEthreader Server
Overview
Methods
Step 1: features preparation.
In this step, an in-house MSA (multiple sequence alignment) detection software is
utilized for collecting homology sequences from UNICLUST, UNIREF90 and METACLUST databases.
The MSA is used for building Henikoff profile, and secondary
structure prediction by PSSpred. The residue-residue contacts of query are predicted by ResPRE for residue pairs with sequence separation of ≥5, as well as residue pairs located at the same helix with a sequence separation of 4. The residue pairs are ranked in descending order of
the confidence scores of the predicted contacts by ResPRE. The top 2.51L residue pairs predicted to be in contacts are selected to form the final contact map of the query.
Step 2: templates detection.
For a given protein with length L, its contact map is an L×L square, binary and symmetric matrix, where residue pairs that are in contacts are designated as 1 and non-contacting residue pairs are set as 0. Eigendecomposition of the contact map is performed, followed by selection of the largest 7 Eigenvalues and corresponding Eigenvectors to calculate the 7-dimensional contact Eigenvector sequences. A scoring function combing contact Eigenvector, secondary structure and profile termss is utilized by semi-global dynamic programming to compare the query sequence with each template in our database. Different templates are then ranked by contact map overlap. Finally, top 5 templates are selected for building full length models.
Step 3: model generation.
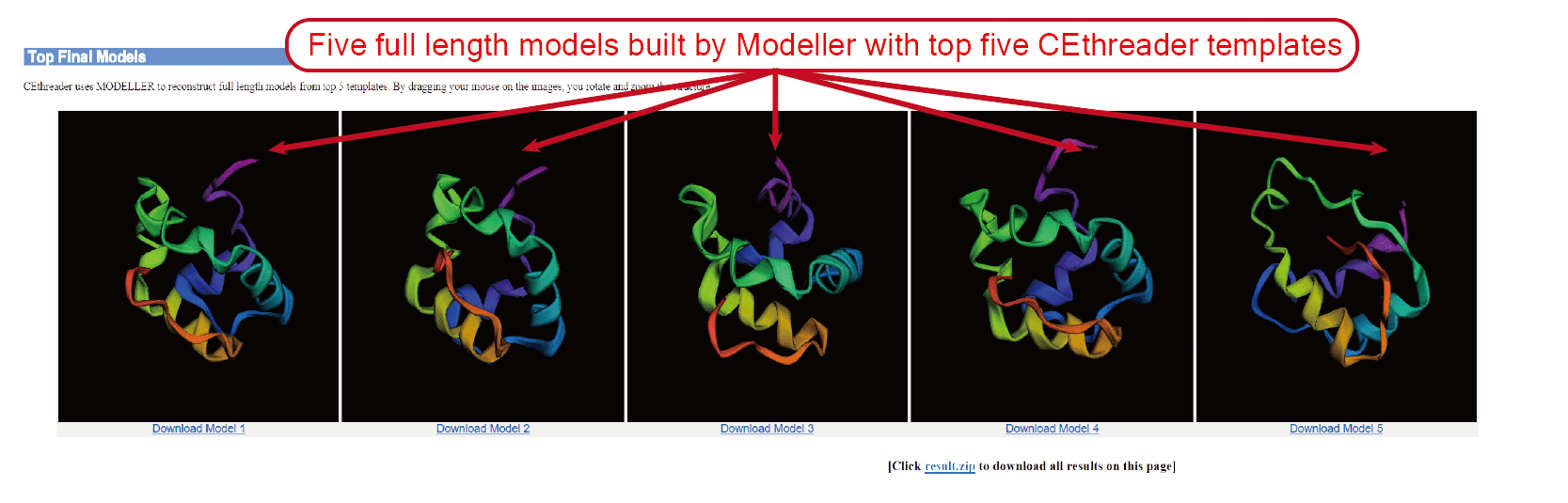
The final models are built by MODELLER with restraints generated from templates and predicted secondary structure.
CEthreader flowchart
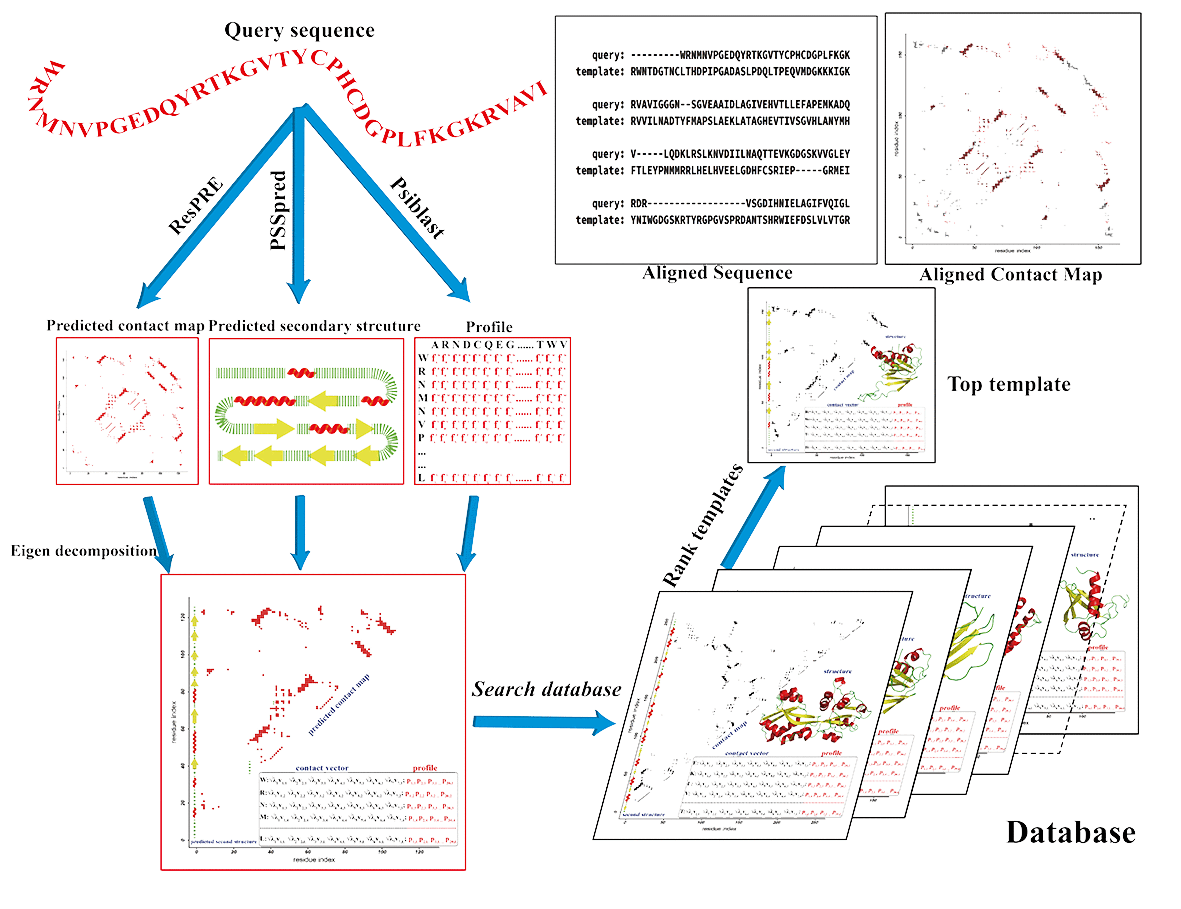
Figure 1. Pipeline of CEthreader.
User Inputs
The user need to paste the FASTA format amino acid sequence to input box, or upload the amino acid sequence of the query protein through browser button.
Figure 2. User inputs.
Content in output page
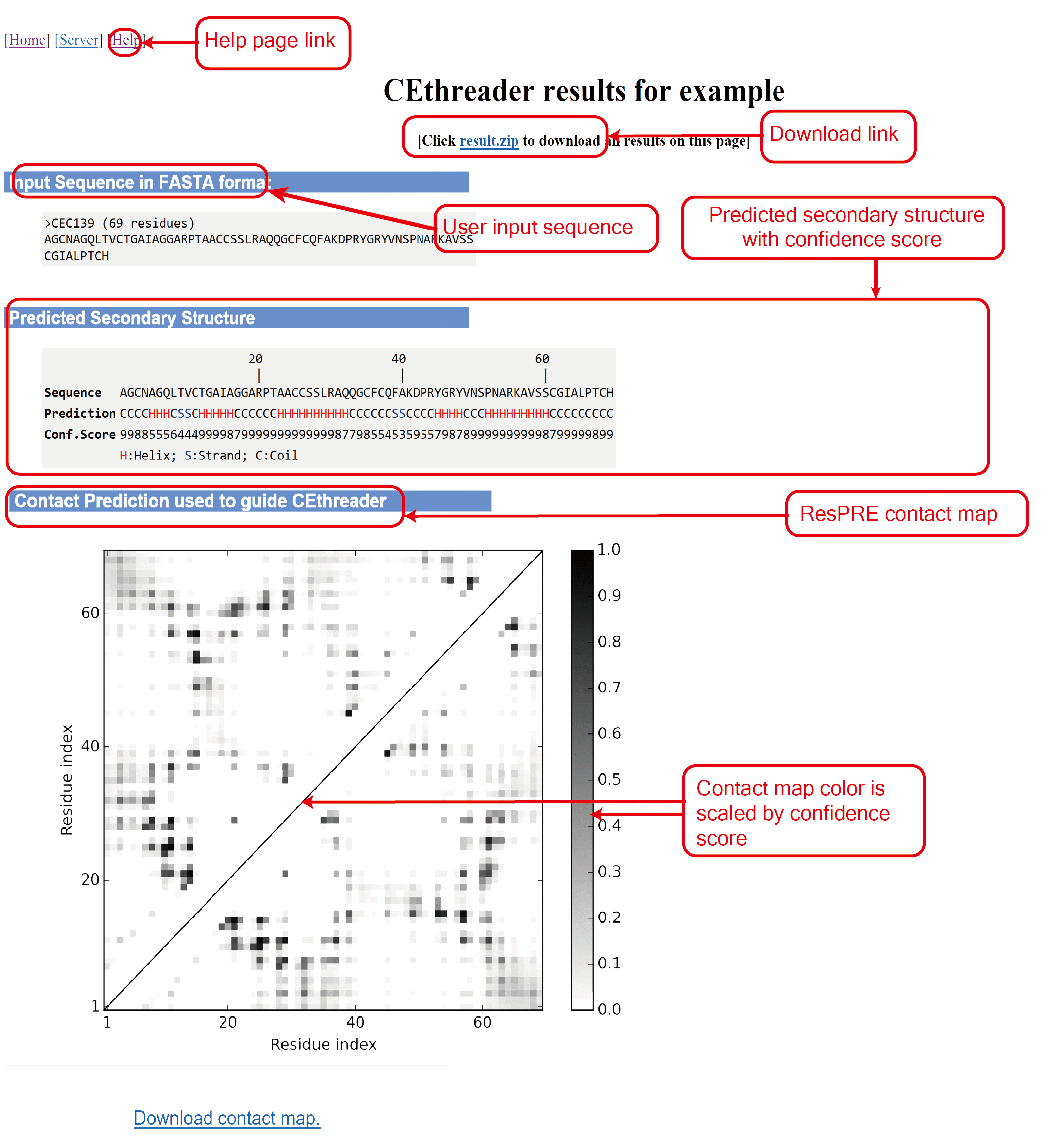
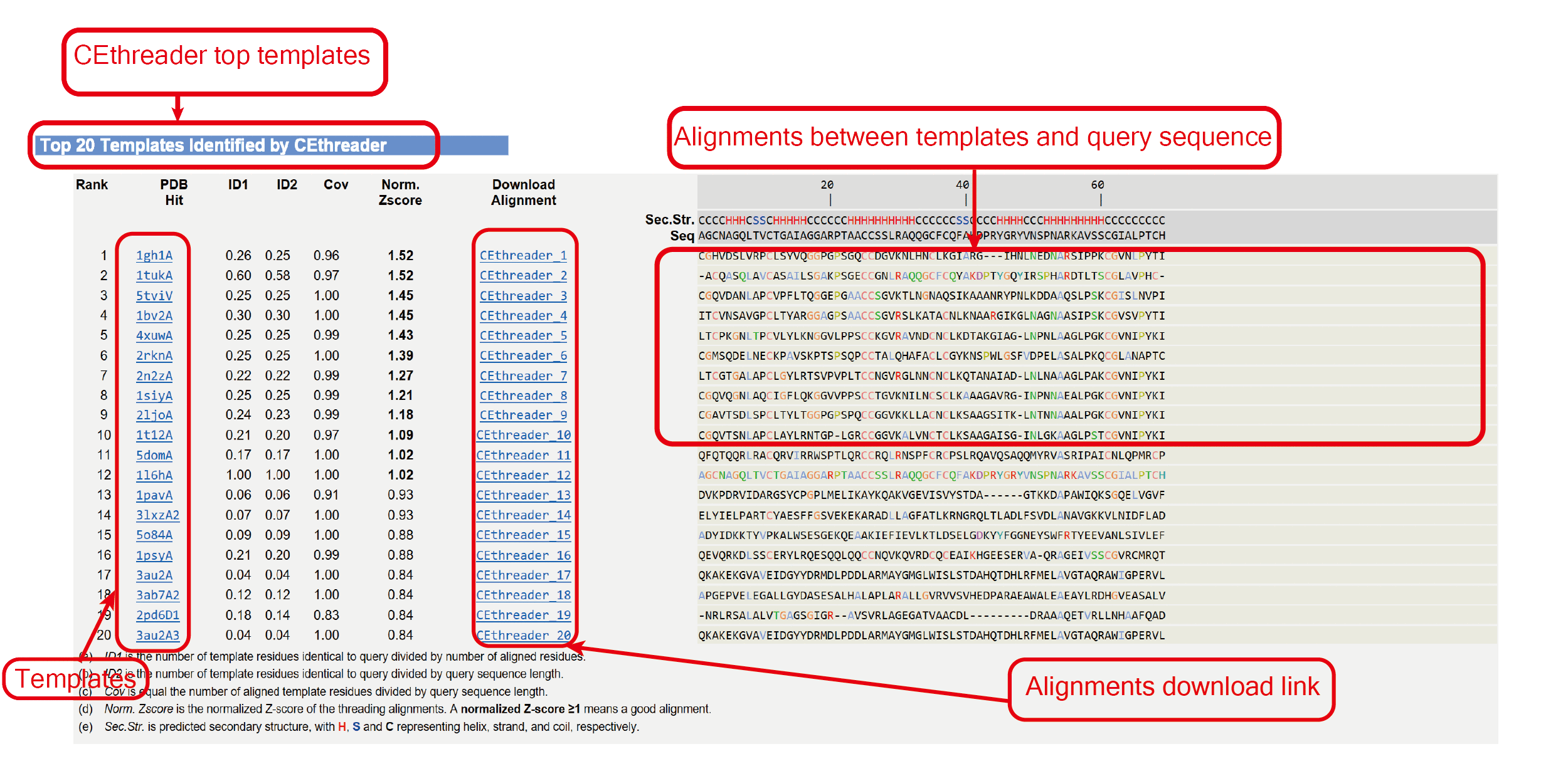
The outputs of CEthreader include:
(i) predicted secondary structure and contact map;
(ii) top 10 templates and associated alignments;
(iii) top 5 full-length models;
Illustration of outputs
Standalone package instruction
References:
Wei Zheng, Qiqige Wuyun, Yang Li, S. M. Mortuza, Chengxin Zhang, Robin Pearce, Jishou Ruan, Yang Zhang.
Detecting distant-homology protein structures by aligning deep neural-network based contact maps. 2019.
Back to the CEthreader homepage